Children’s National Research Institute | Academic Annual Report 2017-2018
Innovation Through Collaboration
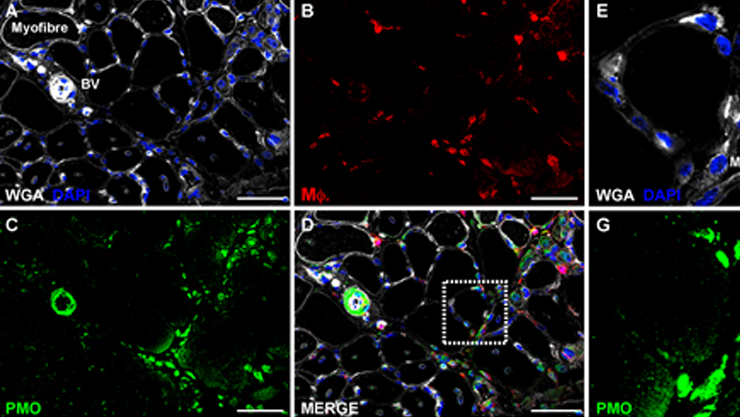
Macrophages and satellite cells mediate phosphorodiamidate morpholino (PMO) antisense delivery to dystrophic muscle
Center for Genetic Medicine Research
Vision: The Center for Genetic Medicine Research seeks to transform children’s health through genome-enabled research, pre-clinical studies of experimental therapeutics, and clinical trials.
The Center for Genetic Medicine Research houses an interdisciplinary faculty, with an almost even distribution of M.D.s and Ph.D.s. Studying health disparities regionally and rare diseases worldwide, faculty and their laboratories create health solutions in personalized and preventive medicine for children. Areas of focus include rare genetic disorders (neuromuscular disorders, leukodystrophies, and urea cycle disorders), airway and lung diseases, childhood brain cancers, and renal diseases. The recruitment of Eric Vilain, M.D., Ph.D., as the new director for the center has expanded the research portfolio to include disorders of reproduction and sexual development and biology of sex differences. Collaboration among faculty members allows many of the center’s projects to incorporate multiple clinical and scientific disciplines. Through a series of National Institutes of Health (NIH) core grants, the center develops and provides access to the latest technologies in genomics, proteomics, microscopy, bioinformatics, preclinical trials, and multi-site clinical trial networks. Center scientists are carving a path for others to follow by developing deep expertise in the emerging areas of rare disease drug development (including personalized medicine), pharmacogenomics, biomarker identification, and acceleration of drug approval. Under the Clark Foundation grant program, the center is providing an infrastructure and $700,000 yearly in pilot funds for Genetic Medicine and CRI faculty to support its mission.
Leadership
-
Eric Vilain, M.D., Ph.D.
Director, Center for Genetic Medicine Research
Faculty List
- Mark Batshaw, M.D. Developmental Pediatrics
- Michael Bukrinsky, Ph.D. Tropical Health, the George Washington University
- Ljubica Caldovic, Ph.D.
- Kimberly A. Chapman, M.D., Ph.D. Rare Disease Institute
- Yi-Wen Chen, DVM, Ph.D.
- Anamaris Colberg-Poley, Ph.D.
- Laurie Conklin, M.D. Gastroenterology, Hepatology and Nutrition (Joint membership with Sheikh Zayed Institute)
- Gary Cunningham, Ph.D.
- Emmanuèle Délot Ph.D. (Joint membership with Center for Translational Science)
- Rohan Fernandes, Ph.D. (Joint membership with Sheikh Zayed Institute)
- Robert J. Freishtat, M.D., MPH Emergency Medicine, Chief of Emergency Medicine
- Alyson Fiorillo, Ph.D.
- Jamie Fraser, M.D., Ph.D. Rare Disease Institute
- Stanley Fricke, Ph.D. Radiology
- Heather Gordish-Dressman, Ph.D. (Joint membership with Center for Translational Science)
- Andrea Gropman, M.D. Neurology
- Lisa Guay-Woodford, M.D. (Joint membership with Center for Translational Science, Director, CTSI-CN)
- Andrea Hahn, M.D. Infectious Disease
- Zhe Han, Ph.D.
- Christopher Heier, Ph.D.
- Jyoti Jaiswal, Ph.D.
- Susan Knoblach, Ph.D.
- Linda Leatherbury, M.D. Cardiology
- Wei Li, Ph.D.
- Hiroki Morizono, Ph.D.
- Evan Nadler, M.D. Surgery (Joint membership with Sheikh Zayed Institute)
- Javad Nazarian, Ph.D.
- Gustavo Nino, M.D., MSHS Pulmonary and Sleep Medicine
- James Novak, Ph.D.
- Aswini Panigrahi, Ph.D.
- Terence A. Partridge, Ph.D.
- Maria T. Pena, M.D. Otolaryngology
- Jason Patregnani, M.D. Cardiology/Critical Care
- Dinesh Pillai, M.D. Pulmonary Medicine
- Hans George Pohl, M.D. Urology
- Diego Preciado, M.D. Otolaryngology (Joint membership with Sheikh Zayed Institute)
- Patricio Ray, M.D.
- Matthew Sharron, M.D. Critical Care
- Dashuang Shi, Ph.D.
- Christopher Spurney, M.D. Cardiology
- Marshall Summar, M.D. (Joint membership with Rare Disease Institute, Director, Rare Disease Institute)
- Mathula Thangarajh, M.D., Ph.D. Neurology
- Laura L. Tosi, M.D. Orthopaedics
- Mendel Tuchman, M.D. Genetics and Metabolism
- John van den Anker, M.D. Pediatric Clinical Pharmacology (Joint with Center for Translational Science)
- Yuan Zhu, Ph.D. (Joint membership with Center for Cancer and Immunology)
Center Research Programs
Gender-Based Biology
Etiology of Disorders/Differences of Sex Development
- Eric Vilain, M.D., Ph.D.
- Hayk Barseghyan, Ph.D.
- Emmanuèle Délot, Ph.D.
- Abhinav Parivesh, Ph.D.
Disorders/Differences of Sex Development (DSD) are a spectrum of developmental conditions with complex overlapping phenotypes placing affected individuals at risk for life-threatening hormonal crises, infertility, cancer, sexual dysfunction, gender dysphoria, and psychosocial distress. While at least 75 genes are known to cause these conditions in humans, each explains a small number of cases, and a majority of patients remain undiagnosed. The team has used animal and cellular models, through candidate gene or genome-wide approaches, to identify new genes and validate novel pathogenic variants for several DSD conditions. The team identified 15 novel candidate genes and demonstrated that they are expressed in a sex-specific manner in the developing gonad, many under the control of the well-known sex-determining gene Sox9 (Barseghyan et al. 2018). To further validate these candidate genes, the team has now undertaken to clone variants of unknown clinical significance found in the genomes of patients with DSD into expression vectors to assay their effects on function of the proteins in vitro. The team is also developing a model of testicular (Sertoli) cell in culture using transdifferentiation to test gene function and variants directly in patient-derived cells.
Neural Stem Cells and Brain Sex Differences
- Eric Vilain, M.D., Ph.D.
- Matthew Bramble, Ph.D.
The laboratory is interested in exploring the biological bases for sex differences in health and disease, in particular what leads to sex differences in brain and behavior. While hormonal influences are well established, genetic diversity and modulation of epigenetic signatures in glia are emerging as mechanisms supporting the establishment of differences between males and females. The laboratory had demonstrated that sexually dimorphic expression of a set of genes in the brains of mouse embryos precedes the making any sex hormones by the embryonic gonad. The lab hasdeveloped a model of mouse neural stem cells to test how these sex differences may be established during development. Using RNA-Seq, the lab has identified ~100 genes that are differentially expressed in XX and XY neural stem cells. The team further showed that neural stem cells respond to androgens in a sex-dependent fashion. For example, when XX cells are treated with testosterone, about 40 percent of the differentially expressed genes acquire a male pattern of expression. Furthermore, testosterone exposure causes a global reduction in DNA methylation, which persists even after testosterone is removed, indicating a possible new reprogramming role for androgens within the developing central nervous system. The team continues exploring these mechanisms as a basis to understanding behavioral and cognitive sex differences in humans, including patients with DSD.
DSD-Translational Research Network
- Emmanuèle Délot, Ph.D.
- Eric Vilain, M.D., Ph.D.
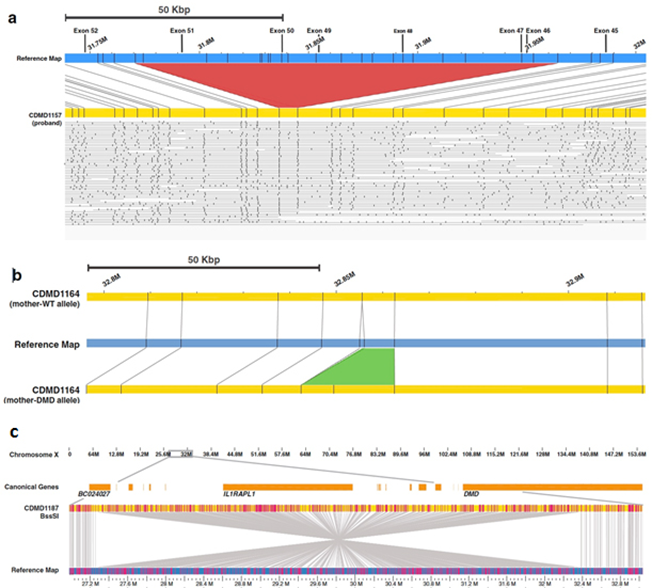
Whole-Genome Mapping identifies pathogenic deletions in Duchenne Muscular Dystrophy patients (Barseghyan et al., Genome Medicine, 2017). a: Fluorescently labeled sites along ultra-long molecules of DNA allow de novo assembly of the proband’s genome (in yellow). Difference in pattern with the reference map (in blue) demonstrates the presence of a multi-exon deletion (red triangle) in the dystrophin gene. b: The mother of a DMD patient carries a 13 kbp insertion on one of her alleles, a structural variant too small to be reportable by clinical chromosomal microarray methods. c: A 5.1 Mbp inversion interrupts the DMD gene. Such a balanced structural variant could not be identified with chromosomal microarrays.
To address the lack of available evidence and the controversies in DSD management, in 2011, the lab founded the first (and only) North American network with four sites and a group of patient advocates, Accord Alliance. The alliance has now expanded to 12 sites, including Children’s National Hospital, and created a unique infrastructure comprising of a registry and a biobank (both of which are now hosted at Children’s National). The team organizes network-wide clinical case conferences to discuss challenging cases. The team created an entire set of clinical forms tailored for DSD patients, in all specialties concerned in this interdisciplinary model of care (endocrine, genetics, psychosocial, urology, gynecology etc.). The registry longitudinally collects the standardized deep phenotyping (including psychosocial), genotyping, as well as parameters of clinical care, allowing for outcomes research. The scientests recently published the principles governing the network and the first analysis of genetic registry data (Délot et al. 2017), as well as a detailed survey of the state of DSD clinical practice across the U.S. (Rolston et al. 2017). The goals of the DSD-TRN are to: 1) determine links between intervention and health outcomes; 2) establish best practice guidelines based on this evidence; 3) educate both providers and patients to these new standards of care; and 4) pilot integration of those standardized clinical forms into Electronic Medical Records, to streamline the clinical workflow and generalize the best practices the team benchmarks, in this disease-specific precision medicine effort.
Steroid Drug Development and Inflammatory Bowel Disease
Dissociative Steroid Drug Development
- Laurie Conklin, M.D.
- Christopher Heier, Ph.D.
- John van den Anker, M.D., Ph.D.
- Christopher Spurney, M.D.
- Robert J. Freishtat, M.D.
- Heather Gordish-Dressman, Ph.D.
Drs. Nagaraju and Hoffman (prior directors of the Center for Genetic Medicine Research) worked with medicinal chemist John McCall to develop dissociative steroids, a new series of drugs that are able to improve the efficacy and decrease the side effects associated with glucocorticoid drugs. The team created a technology transfer company, ReveraGen BioPharma, Inc. (the first for-profit spin-off from Children’s National) and developed the lead compound VBP15 (Vamorolone). ReveraGen developed the drug for use in patients with Duchenne muscular dystrophy (DMD) in collaboration with NIH Therapeutics for Rare and Neglected Diseases (TRND) and with financial support from five nonprofit foundations: Muscular Dystrophy Association (USA), Joining Jack (UK), DRF (UK), Duchenne Children’s Trust (UK), and Parent Project Muscular Dystrophy (USA). Although VBP15’s origins are in the treatment of DMD, the center, working with ReveraGen, has received NIH (STTR) funding to assess efficacy of vamorolone in asthma, sickle cell disease, rheumatoid arthritis, multiple sclerosis, and inflammatory bowel disease (IBD) models.
Phase 1 clinical trials have been successfully completed in about 80 adult volunteers (Hoffman et al. 2018.) Phase 2a studies in DMD children started in July 2016 and are ongoing. ReveraGen has worked with Newcastle University in the United Kingdom to obtain a prestigious European Union grant (Horizons 2020) to support DMD trials. Recently, two papers were published and a U54 Center Grant awarded ($4.3 M from NIH / NICHD- PI, Dr. van den Anker) to develop pharmacodynamic biomarkers for dissociative steroids, as well as other steroids and other anti-inflammatory drugs. These biomarkers will enable investigators to sensitively detect responses to treatment in patients with DMD or (IBD), simply by testing patient blood samples.
Inflammatory Bowel Disease (IBD)
- Laurie Conklin, M.D.
- Christopher Heier, Ph.D.
- John van den Anker, M.D., Ph.D.
Inflammatory bowel disease (Crohn’s disease, ulcerative colitis) affects more than 1.4 million Americans, about one in four of whom are children. Glucocorticoids, such as prednisone, remain one of the most effective and commonly prescribed therapies to induce remission in inflammatory bowel disease. However, lasting side effects, such as growth stunting, hypertension and osteoporosis, limit long-term use. ReveraGen has identified a dissociative steroidal compound, vamorolone (formerly VBP15) which is effective in reducing inflammation without major side effects (Damsker et al. 2016). The team also identified pharmacodynamic biomarkers in serum of patients who respond to treatment with both steroid and biologic drugs (Heier et al. 2016). Further clinical studies, funded by a NIH U54 Center Grant, are under way to bridge these blood-based biomarkers to endoscopic and clinical outcomes. These studies are important steps toward the group’s ultimate goals of improving clinical care and developing Vamorolone as an improved alternative to conventional steroid therapy for patients with IBD.
Airway and Lung Diseases
GenMed’s Airway Biology research group focuses on the “united airway” concept that epithelium and epithelial responses in the respiratory tract are similar and interrelated and that complex interactions between the epithelium and mesenchyme mediate lung development and inflammatory airway diseases. This year saw the publication of key findings advancing clinical care and major new grants ($3 million), including the renewal of the NHLBI-funded K12 (career development) Program in Omics of Pediatric Lung Diseases in DC and the recruitment of two K12 scholars, whose research focuses on the genomics of microorganisms in the lung. The 18 faculty members of the Airway Biology group, led by internationally recognized leaders (Drs. Freishtat, and Preciado), who worked alongside investigators from the Center for Translational Science, the Sheikh Zayed Institute, private industry, and other GenMed scientists. The team studies asthma, cystic fibrosis (CF), otitis media (OM), chronic rhinosinusitis (CRS), lung complications of sepsis and neonatal and infant respiratory disorders.
The Cell Culture Core, a key asset in Children’s National fight to treat airway and lung diseases, assists the respiratory biology research community at-large. The core supports studies in respiratory epithelial biology and trains junior faculty, fellows, and students. Notably, recently they have incorporated the use of conditionally reprogrammed cell (CRC) technology to enhance cell growth and lifespan of nasal and bronchial primary cells from infants and young children. This milestone has greatly advanced the core’s ability to perform translational studies for a myriad of respiratory disorders in all ages.
Asthma
- Robert J. Freishtat, M.D., MPH
- Gustavo Nino, M.D., MSHS
- Geovanny Perez, M.D.
- Dinesh Pillai, M.D.
- Stephen Teach, M.D., MPH
Asthma in the United States is considerably more prevalent and severe than 40 years ago, yet the reasons for this are not clear. It remains one of the most significant childhood illnesses, disproportionately affecting urban youth, especially African Americans, who have among the highest asthma-related morbidity and mortality rates of any U.S. racial/ethnic group. The asthma team brings to bear patient-oriented and data-driven research to identify strategies to reduce the health disparities experienced by disadvantaged, urban, and minority youth with asthma.
The airway biology team uses translational and multidisciplinary approaches to asthma research. Dr. Freishtat’s AsthMaP® Project (www.AsthMaPKids.org), funded by the National Institute on Minority Health and Health Disparities (NIMHD) is the basis for this research. Drs. Pillai and Freishtat lead efforts to mine the data-rich AsthMaP®2 project to gain insights into asthma. In addition, the AsthMaP® Project serves as a central resource for the asthma studies in the center. An example of this is a collaboration between Drs. Freishtat and Perez-Losada and the Computational Biology Institute at George Washington University (GW) using a new statistical framework (PathoScope) to accurately and quickly analyze airway DNA/RNA sequences to study the contribution of the microbiome to asthma and lung infections.
Asthma and Obesity: With rates of asthma and obesity increasing, it is critical to identify mechanisms by which obesity impacts asthma. The two epidemics disproportionately affect minority disadvantaged children, many of whom live in the inner city. The District of Columbia ranks third nationally in childhood obesity and has one of the highest asthma prevalence rates in the country. Obesity has been associated with increased asthma symptoms and poor response to asthma therapy. A multidisciplinary team led by Dr. Freishtat and Evan Nadler, M.D., (SZI) continues to lead the field in the study of how excess fat tissue in obese patients drives abnormal processes in other organs like the lungs.
Refractory Asthma: Dr. Pillai developed Children’s first Severe Asthma Clinic for high-risk children identified by their frequent visits to the Emergency Department and hospital admissions for therapy-resistant asthma. This valuable clinical resource aims to turn research into new treatments for therapy-resistant asthma.
Inflammatory Airway Diseases
- Andrea Hahn, M.D.
- Gustavo Nino, M.D., MSHS
- Maria Peña, M.D.
- Geovanny Perez, M.D.
- Dinesh Pillai, M.D.
- Diego Preciado, M.D., Ph.D.
Many of the pediatric respiratory tract diseases the team studies (asthma, CF, chronic rhinosinusitis CRS, and otitis media (OM])) are typically characterized by mucus hypersecretion resulting from bacterial and viral infection and/or inflammatory responses that are somewhat specific to each disease. MUC5AC and MUC5B are the major secretory mucins in the respiratory tract. MUC5AC is expressed in goblet cells in airway epithelium and thus is poised to be one of the first innate immune responders to infection and inflammation. MUC5B, typically restricted to submucosal glands, can be overexpressed in lung diseases. Drs. Preciado and Pena reported that MUC5B is also the predominant mucin in CRS secretions. Dr. Preciado continues functional studies of upregulation of MUC5B mucin by bacteria and cytokines.
Cystic Fibrosis: CF, a recessive genetic disease, is the most prevalent orphan disease in the U.S. Patient morbidity and mortality are mainly due to lung disease. Dr. Hahn and Dr. Freishtat are studying changes in the lung microbiome in cystic fibrosis children receiving antibiotic therapy. Preliminary data suggest that suboptimal antibiotic therapy against a cultured pathogen preferentially targets commensal microbes, leading to a further dominance of the pathogenic organism. The lab’s data also suggest that suboptimal antibiotic therapy is associated with decreased recovery of lung function following a bacterial lung infection.
Respiratory Infections
- Gustavo Nino, M.D., MSHS
- Andrea L. Hahn, M.D.
- Robert J. Freishtat, M.D., MPH
- Geovanny Perez, M.D.
- Dinesh Pillai, M.D.
Respiratory viruses: Rhinovirus and respiratory syncytial virus (RSV) are the most common cause of respiratory infections in children. Drs. Nino, Perez and Freishtat are investigating the mechanisms of disease of these respiratory viruses in infants and young children. In a recently published paper, the team identified that microRNA (miR)-155, a potent driver of interferon TH1 responses is part of the host defense response against rhinovirus. This complements prior observations in which the master TH2 cytokine, Thymic Stromal Lymphopoietin (TSLP), is associated with rhinovirus infections in infants. Dr. Freishtat also has recently identified that infants with rhinovirus and RSV infections have different nasal airway microRNA profiles associated with NFκB signaling. Ongoing work includes efforts to better study viral respiratory illnesses in young children using new clinical scores and novel lung imaging being developed in collaboration with the Quantitative Imaging/Bioengineering Initiative in the Sheikh Zayed Institute.
Investigations on the airway microbiome are steadily increasing the team’s understanding of the balance between pathogenic and protective organisms in a variety of disease processes. Drs. Perez, Perez-Losada, Nino and Hahn use Next-Gen sequencing to define the microbiome of patients with CF and acute rhinovirus infection. Along with Dr. Pillai they determine microbial populations in bronchiolar lavages from CF patients. Drs. Nino, Perez, Hahn and Perez-Losada have characterized the microbiome changes during natural rhinovirus infection in children born severely premature relative to children born full term.
Neonatal and Infant Airway Disorders
- Gustavo Nino, M.D., MSHS
- Geovanny Perez, M.D.
Drs. Nino and Perez are investigating the pathobiology of respiratory disorders in newborns and young children, the number one cause of hospitalization and mortality in this vulnerable age group. The team is particularly interested in understanding why babies born premature are highly susceptible to respiratory disorders. To address this question, the lab employs a variety of state-of-the-art techniques for respiratory phenotyping including age-appropriate multi-language scales and computer-based analysis of lung imaging and neonatal bedside monitors. The team has already identified that premature infants have autonomic dysfunction leading to acute life-threatening events and airway immune dysregulation (TH2-biased responses) against respiratory viruses causing recurrent hospitalizations. The team also investigating the developmental biology of the respiratory epithelium in human babies using a novel cellular model developed in the lab with conditional reprogramming cell technology to life-extend airway epithelial cells derived from newborns and infants born full-term and premature.
Lung-Related Diseases
- Robert J. Freishtat, M.D., MPH
- Gustavo Nino, M.D., MSHS
- Jason Patregnani, M.D.
- Geovanny Perez, M.D.
Dr. Freishtat leads efforts on behalf of NIH-funded multicenter studies of genetic changes in overwhelming infections (sepsis) in children and is developing a new treatment for the complications of sepsis targeting a blood platelet protein. In collaboration with the neonatal intensive care unit and the Division of Materno-Fetal Medicine Dr. Nino and Dr. Perez have investigated the presence of nasal molecular as well as physiological biomarkers of respiratory disease in newborns and prematurely born infants. In a recent publication, the team identified non-invasive markers of autonomic dysfunction by cardiorespiratory monitoring that may lead to the prevention of respiratory-related hospitalizations in this vulnerable patient population.
Human Cytomegalovirus
- Jyoti K. Jaiswal, Ph.D.
- Anamaris Colberg-Poley, Ph.D.
Drs. Jaiswal and Colberg-Poley study how the human cytomegalovirus (HCMV) reprograms host cell functions to facilitate viral growth and survival. HCMV infection targets a newly characterized sub-organelle, mitochondria-associated membranes (MAM). The MAM is an endoplasmic reticulum (ER) subdomain that contacts mitochondria and allows for inter-organelle crosstalk. The MAM plays a critical role in ER calcium (Ca2+) signaling to mitochondria (needed for cell metabolism), ER stress responses, innate immunity, and programmed cell death.
Work by this group has previously identified that a viral protein called viral mitochondria-localized inhibitor of apoptosis (vMIA) which localizes prominently to the MAM and Mitochondria, and is required for the anti-apoptotic role of this protein. Using high-resolution fluorescence microscopy the group studies the mechanisms underlying vMIA trafficking. Using superresolution microscopy, they have found that the viral protein vMIA is organized in nanometric clusters at the outer mitochondrial membrane (OMM), similar to some of the other host mitochondrial proteins, including the translocase of the outer mitochondrial membrane (TOM) and voltage-dependent anion channel (VDAC). Work by the graduate student Mansi Mehta involving genetic and pharmacological inhibition of TOM complex, has helped investigate the requirement of this complex in incorporation of this membrane-anchored protein into the outer mitochondrial membrane. These studies are providing insight into a novel pathway that the virus may have adopted to target the host cell mitochondria and thus protect this protein from the host machinery to keep this protein from localizing to the mitochondria.
Ciliary Dysfunction
- Linda Leatherbury, M.D.
- Iman Sami, M.D.
Dr. Leatherbury (cardiology) and Dr. Sami (pulmonary) have started a “Ciliary Dysfunction in Congenital Heart Disease and Suspected Primary Ciliary Dyskinesia” multidisciplinary clinic for research protocol patients. The team and Dr. A. Koumbourlis (Pulmonary Medicine) are co-investigators with Cecilia Lo, Ph.D. (University of Pittsburgh), on a grant from the Department of Defense (DOD) to study pulmonary function in children with congenital heart diseases.
Muscle Biology and Diseases
Cell Biology of Myogenesis, Muscle Repair, Regeneration, and Aging
- Terence Partridge, Ph.D.
- Jyoti Jaiswal, Ph.D.
- James Novak, Ph.D.
Dr. Jaiswal’s group focuses on understanding the cell biology of muscle and degenerative diseases. The group studies the cellular and molecular mechanisms involved in subcellular trafficking and the role played by this process in healing the injured cell membrane and identifying therapies to target diseases resulting from poor repair of injured muscles. Limb Girdle Muscular Dystrophies encompass diseases where ability of the wounded muscle cells to repair is compromised. These studies suggest a role of acid sphingomyelinase and a modified steroid drug – vamorolone, as a potential therapy for LGMD2B. In preclinical studies by Sen Chandra Sreetama, Ph.D., is providing evidence that use of these drugs improve myofiber repair, reduce muscle inflammation and prevent adipogenic conversion of muscle. It has also helped identify a novel role for mitochondria - facilitating repair of injured muscle fibers, deficit in this contributes to myofiber necrosis in Duchenne Muscular Dystrophy (Vila et al 2017). Extending this work further, Adam Horn has identified the molecular mechanism by which mitochondria sense and help repair myofiber injury, work was recognized as one of the top ten amongst the 850 articles published this year by Children’s National investigators (Horn et al 2017). Pursuing a career-development award funded by the Muscular Dystrophy Association Dr. Marshall Hogarth’s work is providing novel clues regarding how poor myofiber repair and chronic inflammation in LGMD2B is linked to adipogenic replacement of the muscle fibers, and has provided evidence supporting a role of inflammation in adipogenic replacement of muscle in LGMD2B (Defour et al 2017).
Work by Marie Nearing and Davi Mazala in Dr. Partridge’s lab are investigating the differences in the mechanisms of muscle formation during the growth period immediately after birth from those that operate during subsequent regeneration in response to damage caused by muscular dystrophy. The team is exploring how the mechanisms that control satellite cell function during growth differ from those that operate during regeneration, and testing the responses of muscle to growth-promoting agents, such as Formoterol. These investigations are carried out in the mdx mouse, a model for human DMD. Understanding of these processes is important, because in boys with Duchenne, unlike mice, growth and repair mechanisms operate contemporaneously for much of the juvenile and pubertal growth period. It was recently shown that the DMD mutation on the DBA/2J mouse background results in an atrophic phenotype that more closely resembles that in DMD boys. Dr. Davi Mazala, together with Dr. Novak and Dr. Jaiswal, is now investigating the pathological differences between the DBA/2J model and the original mdx mouse, to identify the crucial factors underlying the distinct pathologies and refine the utility of the DBA/2J model for testing of therapeutic strategies such as exon-skipping.
Investigation of Antisense Oligonucleotide Delivery and Pharmacokinetics
- James Novak, Ph.D.
- Terence Partridge, Ph.D.
- Jyoti Jaiswal, Ph.D.
- John N. van den Anker, M.D., Ph.D.
The use of splice-switching antisense oligonucleotides (ASO) for genetic diseases like DMD has moved from the bench to the clinic. Despite this recent success, efficacy of ASO delivery has remained far from optimal, limiting the extent of clinical benefit of ASO-based therapies for DMD and other diseases. The laboratory of Dr. Novak, together with the laboratories of Dr. Partridge and Dr. Jaiswal, has recently identified tissue inflammation as an unexpected ally for overcoming this constraint. Working to understand the mechanism of ASO delivery in DMD, the team discovered that inflammatory cells function as an intramuscular reservoir that locally stores the intravenously administered ASO in regions of muscle necrosis and repair (Novak et al. 2017). This finding points to a potentially novel strategy for systemic ASO delivery, involving the use of inflammatory cells as a Trojan horse. Such an approach would have the benefit not only of enhancing tissue-specific delivery of ASO, but also of reducing the impact of their rapid clearance from the circulation. The team is also investigating 1) ASO delivery strategies for multi-exon skipping with the DMD exon 45-55 ‘hot spot’ region; 2) mechanisms regulating the delivery of cell-penetrating peptide (CPP) antisense agents; 3) long-term consequences of chronic morpholino exposure to muscle and kidneys; and 4) surrogate biomarkers as predictive outcome measures of morpholino-induced exon skipping at distinct periods of growth or heightened pathology in mouse models of the disease. This research is supported by a NIH U54 grant on pediatric pharmacology, Parent Project Muscular Dystrophy, and Early Stage Investigator Development Grants awarded to Dr. Novak through the M.D.A., Duchenne Parent Project – Netherlands, and Foundation to Eradiate Duchenne.
Pathophysiological Mechanisms and Therapies for Muscular Dystrophies
- Yi-Wen Chen, DVM, Ph.D.
The team uses omics approaches to dissect disease mechanisms and identify circulating biomarkers for muscular dystrophies and autoimmune muscle disorders. Among the diseases studied, the lab been most interested in facioscapulohumeral muscular dystrophy (FSHD) which is caused by both genetic and epigenetic mechanisms. After the double homebox protein 4 (DUX4) was identified as the causative gene of FSHD, the team has been studying molecular mechanisms that regulate DUX4. Recently, the scientests shifted research focus to therapeutic development and preparation for clinical trials. In collaboration with Jean Mah, M.D., and the Cooperative International Neuromuscular Research Group (CINRG), the lab conducted a natural history study of early onset FSHD and collected blood samples for biomarker studies using proteomes profiling and RNA-seq. For therapeutic development, the lab is investigating antisense oligonucleotide strategies for knocking down DUX4 in muscle cells using both cell and mouse models. A provisional patent has been filed for one of the compounds which are under investigation. Accurate and fast diagnosis of disease conditions is critical for successful disease interventions. FSHD is caused by contraction of 3.3 kb D4Z4 repeat, which posed technical challenges to use conventional molecular tools for molecular diagnosis. The team is currently developing a long-read sequencing assay for diagnosis and research using Nanopore MinION.
Development of Improved Treatments for Muscle Diseases
- Christopher Heier, Ph.D.
- Laurie Conklin, M.D.
Our goal is to develop next generation treatments and biomarkers for diseases in children. By targeting the underlying molecular biology of muscle diseases the team believes it can help a broad array of human illnesses. For example, the lab is currently dissecting the molecular signaling pathways of steroids in DMD. This helped develop a new anti-inflammatory drug named vamorolone to the point that it is now in Phase 2 clinical trials for DMD. Early work indicates this drug has the potential to replace prednisone as the standard of care for DMD, by providing a drug that is more effective while also being much safer. Ultimately, this can greatly improve treatment of DMD as well as a number of other diseases with shared molecular biology.
Improving Heart failure and Fibrosis in Children
- Christopher Heier, Ph.D.
- Christopher Spurney, M.D.
Heart failure is a leading cause of death in DMD, and is a feature of several other genetic muscle diseases. Currently, Drs. Heier and Spurney are testing a new anti-inflammatory drug, vamorolone, for children with DMD. They find that this drug also possesses a new property that improves heart failure and fibrosis. By acting as a mineralocorticoid receptor antagonist, vamorolone effectively protects hearts and improves blood pressure in animal models. These data are consistent with clinical findings that another mineralocorticoid antagonist (eplerenone) improves heart failure in DMD patients. Moving forward, they will further investigate this new drug mechanism, determine the potential of vamorolone to treat heart failure in other diseases, and identify blood-based biomarkers that predict heart health and the need for interventions.
Using MicroRNA to Improve Treatment of Childhood Diseases and Chronic Inflammation
- Alyson Fiorillo, Ph.D.
- Christopher Heier, Ph.D.
- Laurie Conklin, M.D.
A new area of research, microRNA, is emerging as an important new source of biomarkers and therapeutic targets. We find a set of microRNAs are increased during states of inflammation, and conserved across several diseases featuring inflammation. By measuring levels of these microRNAs in blood, we find we can develop new minimally invasive measures that predict disease and drug response in children. Additionally, these same microRNAs are conserved in animal models of disease; we find that they can help guide and improve decisions on when to move a drug from preclinical animal testing into human clinical trials. Moving forward, we are developing develop next generation drugs that target these same microRNAs with high specificity in hopes to produce improved treatments for chronic inflammation. Our current focus is on DMD and inflammatory bowel disease, though we have supportive data on myositis and BMD as well. These microRNA studies are helping to develop new therapies, as well as new methods to track the effective treatment of patients.
The Role of MicroRNAs in Dystrophin Regulation and Dystrophin Disease Progression
- Alyson Fiorillo, Ph.D.
The Fiorillo lab has a strong focus on DMD where it previously identified seven microRNAs that bind to the dystrophin 3’UTR and inhibit dystrophin translation (termed dystrophin-targeting microRNAs or DTMs). These microRNAs are regulated by NFkB-mediated inflammatory signaling, are highly upregulated in DMD muscle, and are detrimental to the success of dystrophin rescue in DMD muscle via exon skipping (Fiorillo et. al 2015). Building on previous work the current goals of the Fiorillo Laboratory are to: 1) dissect the mechanisms and consequences of microRNA dysregulation in DMD and other inflammatory muscle disorders; 2) utilize mouse models to determine proof-of-concept of microRNA or oligonucleotide-based therapeutics; 3) investigate why diseased/damaged muscle begins to take on an immune cell-like signature; and 4) explore how the crosstalk between muscle and immune cells promotes a chronic inflammatory state.
Clinical Trials and Cooperative International Neuromuscular Research Group (CINRG)
- Heather Gordish-Dressman, Ph.D.
CINRG (CINRGresearch.org) is a consortium of medical and scientific investigators from academic and research centers who share the common goal of improving the lives of patients with neuromuscular disease and their families. The CINRG network joins together over 25 clinical and research sites from around the world to perform clinical studies in neuromuscular disorders. The group has successfully enrolled more than 1,200 study participants (predominantly children) into 18 studies to date. Dr. Gordish-Dressman serves as the principal investigator (PI) at Children’s National for both the CINRG Duchenne and Becker Natural History Studies.
The Duchenne Natural History Study (DNHS), chaired by CINRG PI Craig McDonald, M.D., of UC-Davis, has completed funding through a combination of government (National Institute on Disability and Rehabilitation Research at NIH and DOD), foundation (Parent Project Muscular Dystrophy), and industry partner grants. It is the largest natural history study of DMD to date, with a wealth of data that provide natural history controls for both the design of industry trials and the interpretation of clinical trial data for many clinical and biochemical endpoints. The value of these data is recognized in the muscular dystrophy research community globally, resulting in increased interest in confidential access to the data. Dr. Gordish-Dressman continues this effort as lead statistician and has authored several publications in 2017 including a seminal manuscript published by the Lancet in November 2017.
The Becker natural history study, is led by PI Paula Clemens, M.D., in Pittsburgh. The study has reached its enrollment goals and is collecting long-term data on participants with Becker muscular dystrophy. The CoQ-10/Lisinopril clinical trial, funded by the DOD and led by Dr. Clemens is continuing with long-term follow-up of participants. Dr. Gordish-Dressman is currently analyzing data from both of these important studies.
Urea Cycle Disorders (UCD)
- Mendel Tuchman, M.D.
- Mark Batshaw, M.D.
- Nicholas Ah Mew, M.D.
- Ljubica Caldovic, Ph.D.
- Andrea Gropman, M.D.
- Hiroki Morizono, Ph.D.
- Dashuang Shi, Ph.D.
The Urea Cycle Disorders Consortium (UCDC)
The UCDC is an NIH-funded 16-site research consortium within the Rare Disease Clinical Research Network to investigate inborn errors of the urea cycle. These rare genetic disorders result from defects in any of the eight genes associated with this important metabolic cycle and have a combined prevalence of about 1in 30,000. Urea cycle disorders (UCDs) lead to the accumulation of ammonia in the blood and brain and resultant episodes of metabolic encephalopathy, with a great risk of morbidity and mortality. The focus of the UCDC is to perform a longitudinal natural history study and intervention studies of these disorders and to develop and test new diagnostic and therapeutic approaches. Children’s National serves as the leadership hub of the consortium, which is led by Dr. Gropman. The UCDC is supported by funding from the NIH and the Kettering and O’Malley Family Foundations. In the past decade, the consortium successfully brought to market three new drugs to treat hyperammonemia and currently follows more than 700 individuals with these disorders.
Neuroimaging in Urea Cycle Disorders
Advanced neuroimaging technology, using diffusion tensor imaging, volumetric averaging, fMRI, and magnetic resonance spectroscopy, allows non-invasive investigations of the brain in complex conditions such as hyperammonemia in UCDs. Dr. Gropman and her team, including John VanMeter, Ph.D. (Georgetown University), and Drs. Whitehead and Fricke, have been using these methods to identify biomarkers that reflect the downstream impact of UCDs on cognition. Previous imaging research performed as part of the UCDC identified specific biomarkers of neurologic injury in ornithine transcarbamylase deficiency (OTCD). Specifically, the study showed that elevations in brain glutamine, a storage depot for ammonia, may persist and be associated with alterations in mental status and cognition even in the presence of normal plasma ammonia and normal or only slightly elevated plasma glutamine. In addition, another small biomarker, myoinositol, may be associated with cognitive reserve in patients who have had hyperammonemia. Female carriers of OTCD, an X-linked UCD, who are expected to have milder symptoms, demonstrate challenges in executive function and working memory, although they may function well with simple tasks. This was shown by performance on a number of cognitive tests that target frontal lobe function and by activation and resting state studies on fMRI. While characterization of mutations can be achieved in most cases, this information does not necessarily predict the severity of the underlying neurological compromise in patients. The clinical phenotype varies from one patient to another and results in significant outcome heterogeneity. The group’s neuroimaging studies revealed affected cognitive domains, which include nonverbal learning, fine motor processing, reaction time, visual memory, attention, and executive function. Deficits in these capacities may be seen in symptomatic patients, as well as in asymptomatic carriers with normal IQ, and correlate with variances in brain structure and function in these patients.
These studies allow the team to begin to understand the brain pathophysiology in hyperammonemia and correlate the results with different variables, including treatment modalities. Current studies are aimed at understanding the chronology of recovery from hyperammonemia using neuroimaging biomarkers and studying the brain effects of other UCDs besides OTCD. Dr. Gropman’s group is also exploring the use of optical imaging as a totally non-invasive technique to target the very young and more cognitively challenged patients with OTCD.
Clinical Trials in Hyperammonemia
In an NIH-funded project, Drs. Tuchman and Ah Mew demonstrated that an oral medication, N-carbamylglutamate, can correct the biochemical defect in patients with a UCD known as N-acetylglutamate synthase (NAGS) deficiency, thereby normalizing ammonia levels and restoring normal urea production. Results from this study led to the discovery of the first regulatory mutation in the NAGS gene. Subsequent clinical studies showed that N-carbamylglutamate can reduce ammonia levels and improve urea production in patients with other forms of hyperammonemia, such as carbamyl phosphate synthetase (CPS1) deficiency, and propionic and methylmalonic acidemia. The success of this translational work has led to an NIH-funded groundbreaking randomized, double blind, placebo-controlled clinical trial of N-carbamylglutamate in patients with the aforementioned disorders who present with acute hyperammonemia. Results from this trial should be available within the next year and could potentially be used to expand the clinical indications for this drug.
Liver Transplant in the Urea Cycle Disorders
In a project funded by the Patient-Centered Outcomes Research Institute (PCORI), Drs. Ah Mew and McCarter are conducting a study to compare the outcome of liver transplantation with conservative management in urea cycle disorders. This work is being done in collaboration with the School of Public Health at GW, the National Urea Cycle Disorders Foundation, and the Emmes Corporation’s Studies of Pediatric Liver Transplantation. The results of the study should be available at the end of 2018 and will greatly help future UCD patients and providers through the difficult process of deciding whether or not to proceed with liver transplantation.
Neuroprotection from Hyperammonemia
In addition to ammonia removal, a new therapeutic paradigm for treating hyperammonemia directly protects the brain. Drs. Caldovic, Tuchman, and Morizono are screening chemicals as potential drugs that can protect the brain from the toxic effects of ammonia. In a project previously funded by NIH and now by industry, the team developed a zebrafish model of hyperammonemia and is using it to screen thousands of chemicals for their ability to prolong survival of zebrafish larvae in water containing high ammonia concentrations. Several chemicals that affect neurotransmission were already documented in this screen to protect zebrafish from high ammonia exposure. Dr. Caldovic received a pilot award to investigate whether chemicals affecting different neurotransmission systems act synergistically to provide more effective neuroprotection. Two pairs of chemicals were more effective at protecting zebrafish than either chemical alone. The team has also identified early behavioral markers of hyperammonemia in mice and has shown that changes in behavior coincide with the onset of abnormal neuronal electrical activity in mice experiencing hyperammonemia. This will allow the team to test efficacy of the lead compounds in a mouse model of inducible hyperammonemia. Those confirmed to be both effective and nontoxic will proceed to clinical trials.
Gene Therapy for Urea Cycle Disorders
Drs. Morizono and Batshaw, along with their long-term collaborators at the University of Pennsylvania, James Wilson, M.D., Ph.D., and Lili Wang, Ph.D., have been investigating the efficacy of adeno-associated virus (AAV)–based gene therapy for the treatment of OTC deficiency (the most common urea cycle disorder) in mouse models. The virus is used to deliver a functional copy of the OTC gene to the liver. At the inception of this project, it took more than two weeks for AAV gene delivery to reach protective levels of OTC gene expression, which is a problem for this neonatal-onset, potentially fatal disorder. Continual optimizations of the vector reduced this time from days to hours, resulting in a clinical candidate vector design. The team’s industry partner has begun enrolling adult patients with a milder form of OTC deficiency to participate in a clinical trial using this AAV vector. The preclinical program has also successfully demonstrated that CRISPR/Cas9-based gene editing can correct specific mutations in OTC-deficient mice at levels that protect against a hyperammonemic episode. Drs. Caldovic, Morizono and Tuchman have also shown that AAV-based gene delivery of the NAGS gene successfully rescues NAGS-deficient mice so they do not require N-carbamylglutamate supplementation for survival.
Structural Biology of N-Acetylglutamate Synthase (NAGS) Deficiency
In another project funded by the NIH, Drs. Shi’s and Dr. Caldovic’s laboratories continued to investigate the structural biology of NAGS and CPS1 proteins of the urea cycle. Dr. Shi’s group created an insect cell expression system for both proteins, which allows further structural studies to be conducted. They now understand mechanism for successful treatment of a patient with CPS1 deficiency with N-carbamylglutamate and can personalize use of this drug in similar patients. Dr. Caldovic’s group compared biophysical properties of bacterial and mammalian NAGS proteins, and found, with high resolution crystal structures, that some NAGS proteins are ensembles of different oligomers while others are not. This result provides rationale for selecting dog, Tasmanian devil, American bison and domestic yak NAGS proteins as good candidates for structural studies of the whole protein.
Regulation of Ureagenesis by NAGS
In a project funded by the NIH, Drs. Tuchman, Caldovic and Morizono created a mouse model with complete NAGS deficiency that can be rescued by supplementation of N-carbamylglutamate and L-citrulline. This is the only mouse model of a urea cycle defect that can be rescued to reach adulthood and reproduce. The team has now used this model and adeno-associated virus (AAV)–based gene transfer to investigate the function of NAGS in vivo. Delivery of E354A arginine-insensitive NAGS via AAV-based gene transfer system rescued the NAGS deficient mice but they had elevated plasma ammonia. Dr. Caldovic’s team also carried out biochemical and biophysical characterization of an arginine-insensitive NAGS protein that was found in a patient with NAGS deficiency. This shows that binding of arginine to NAGS is important for its function and efficient ureagenesis. Dr. Caldovic’s laboratory is studying conserved DNA sequences upstream of the NAGS gene and in its first intron that seem to regulate its expression and where mutations (missed by clinical testing) can lead to hyperammonemia. The lab used computational methods and reporter gene assays to show that five sequence variants found outside of the NAGS coding region affect expression of the NAGS gene and lead to NAGS deficiency.
Rare Disease Institute
- Marshall Summar, M.D.
- Gary Cunningham, Ph.D.
Marshall Summar, M.D., and his team work together across a number projects using a team/collaboration approach. This has led to the development of a number of devices for detection of biomarkers of disease in PKU, hyperammonemia, and maple syrup urine disease. These devices were licensed for U.S. Food and Drug Administration approval and manufacture in 2016.
There are a number of therapeutic projects underway. The recombinant enzyme Phenylalanine Dehydrogenase derived from thermostable bacteria has been licensed for rapid development as an oral therapy for PKU. Working with Hemoshear Therapeutics and Dr. Chapman the team has developed a novel use for explanted human livers from patients with inborn errors of metabolism for deep molecular phenotyping and testing. This has led to the identification of targets and chemistries for the treatment of acidemia.
Organic Acidemia
- Kimberly A. Chapman, M.D., Ph.D.
Dr. Chapman is pivotal in the research collaboration with Hemoshear Therapeutics and is the leader and organizer of a multi-center team (Children’s National, Stanford University, University of Utah, University of Pittsburgh, and University of Washington) using explanted livers from patients with inborn errors of metabolism in studying organic acidemias. This work has led to a new avenue of therapeutic investigation for patients with organic acidemias and created a bio-repository of cells for six different disorders. Her work in studying the physical structures of the enzymes in organic acid metabolism has led to discovery of interaction between these enzymes and the Krebs cycle. The newly described finding helps explain many of the clinical puzzles in energy metabolism in these patients.
Modeling Pathogenesis in Inborn Error of Metabolism
- Jamie Fraser, M.D., Ph.D.
Dr. Fraser joined the team in 2016 from the NIH. Her specialty is the effects of intermediary metabolism defects on brain function and injury. She has developed a line of embryonic stem cells from mice with inborn errors of metabolism (IEMs), from which are derived cerebral and deep brain organoids. Using this model, along with animal models of IEMs, she is using MRI spectroscopy and deep biochemical analysis to study the molecular consequences of these diseases. This work should prove highly translatable to more widespread models of neurologic stress/damage.
Brain and Spinal Cord Disorders
Central Nervous System (CNS) Injury and Neurodegenerative Disease
- Susan Knoblach, Ph.D.
Dr. Knoblach research focuses on mechanisms of neurodegeneration. Together with scientists in France and Ireland, her lab represents the VITAL (Vesicles in Transmission of ALS) Consortium, which is supported by TARGET ALS. The goal of the consortium is to determine the role of secreted vesicles, known as exosomes, in the progression of amyotrophic lateral sclerosis (ALS). In the past year, using state-of-the-art genomics and proteomics methods, the group characterized the types of proteins, mRNA/miRNAs and miRNAs contained in the vesicles. While exosomes are secreted as part of normal communication between neurons and neurons and muscle, the group found that in ALS, the number of exosomes secreted is markedly high, and that the content of the exosomes is very different from normal. Presently Dr. Knoblach and associates are completing studies to validate candidate proteins/mRNAs in serum from patients with ALS. Some of these may be useful as biomarkers of the disease phenotype (there are 7 distinct clinical types of ALS), stage of disease or response to treatment(s). Other studies are directed at determining how these secreted factors cause or protect against neuronal cell death. In this regard, the lab has identified a group of proteins that activates the so-called FUS cell death pathway, which has previously been associated with ALS, but is not well understood.
Cancer
Childhood Brain Cancers
- Javad Nazarian, Ph.D.
- Suresh Magge, M.D.
- Lindsay Kilburn, M.D.
- Eugene Hwang, M.D.
- Young Kim, M.D.
- Courtney Johnson, M.D.
Despite four decades of clinical trials, diffuse intrinsic pontine glioma (DIPG) remains one of the deadliest childhood cancers. Dr. Nazarian was recently appointed as the scientific director of the Brain Tumor Institute (BTI) at Children’s National. The BTI consists of clinicians and basic researchers who focus on pediatric brain cancers including gliomas and neuroblastoma. Children’s National has recently partnered with the Children’s Brain Tumor Tissue Consortium (CBTTC, Drs. Nazarian and Rood are PIs). These consortia aim to unify the knowledge and resources of member institutions for defining DIPG biology, biomarker identification and discovery of therapeutic targets. The multidisciplinary team of experts includes neurologists, pathologists, neurosurgeons, bioengineers, and oncologists. The Nazarian laboratory is supported by generous funds from the Smashing Walnuts Foundation, Goldwin Foundation, Musella Foundation, Brain Tumor Foundation, Zickler Family Foundation, Mathew Larson Foundation, and Kisses for Kayla Foundation and by the Children’s CTSI.
Drs. Nazarian and Eric Raabe, M.D., Ph.D., (Johns Hopkins School of Medicine) have received funding from the Cure Starts Now for their collaborative and translational work on childhood brain cancers. The team has generated a comprehensive molecular profile (gene, RNA, protein, and microRNA) of pediatric brain stem tumors. In addition, Drs. Hwang and Nazarian are involved in multiple complementary and parallel projects seeking to examine and leverage the immune response against DIPG. One project seeks to evaluate a cytotoxic T-lymphocyte product that will shortly advance to early clinical trials in patients with DIPG.
Dr. Lindsay Kilburn (pediatric oncologist) directs the clinical trial conducted by the Pacific Pediatric Neuro-Oncology Consortium (PNOC), a network of 15 children’s hospitals that conduct clinical trials of new therapies for children with brain tumors. PNOC members have formed a single arm multicenter feasibility trial to use a new treatment approach based on each patient’s tumor genomic profiling consisting of whole exome sequencing (WES) and RNA sequencing (RNA seq) as well as predictive modeling.
Genetic Targets for Acute Myeloid Leukemia
- Zhe Han, Ph.D.
- Jun-yi Zhu, Ph.D.
- Xiaohu Shawn Huang, Ph.D.
- Yiming Ma, Ph.D.
Acute Myeloid Leukemia (AML) is the most difficult type of leukemia for effective treatment, mainly because many genetic mutations could cause AML and their disease mechanism could be very different. Using the powerful genetic tools in Drosophila, the Han lab has developed a series of personalized Drosophila AML models using the oncogenic human genes carrying the exact same mutations from patients. The team has performed genetic screen and drug screen using these fly leukemia models to identify therapeutic targets and potential drug treatment.
Kidney Disease
Polycystic Kidney Disease
- Lisa Guay-Woodford, M.D.
Dr. Guay-Woodford’s major research effort focuses on identifying the clinical and genetic factors involved in the pathogenesis of autosomal recessive polycystic kidney disease (ARPKD). This work has three components: 1) executing an NIH P30-funded effort to extend the ARPKD clinical database and expand the companion biorepositories (DNA and tissue); 2) characterizing disease-causing genes and performing complex trait analyses to identify candidate modifier genes in recessive PKD; and 3) establishing clinical guidelines for optimizing the care of ARPKD patients. As part of the International ARPKD Consortium, her group has cloned PKHD1, the major gene involved in human ARPKD.
In addition, she has characterized two distinct mouse models, cpk and bpk, in which the disease phenotype closely resembles human ARPKD and identified the genes, Cys1 and Bicc1, which are disrupted in each model, respectively. The laboratory’s current efforts are centered on characterizing the functional roles of these recessive PKD genes, their protein products, and the genetic modifiers in normal development and disease pathogenesis.
HIV-1 Associated Renal Diseases
- Patricio E. Ray, M.D.
- Jharna Das, Ph.D.
- Jinliang Li, Ph.D.
- Xuefang Xie, Ph.D.
- Pingtao Tang, Ph.D.
- Sofia Perazzo, M.D.
HIV-associated nephropathy (HIVAN) is a renal disease almost exclusively seen in people of African ancestry. More than 2 million HIV-infected children living in sub-Saharan Africa are at high risk of developing HIVAN if they do not receive adequate anti-retroviral therapy (ART). HIVAN is characterized by the collapse of glomerular capillaries and microcystic transformation of renal tubules, leading to rapid chronic renal failure. These changes are caused by the infection of podocytes and renal tubular epithelial cells (RTEc), yet the mechanism is unclear. Two genetic risk variants in the human APOL1 gene (G1/G2) were identified as major risk factors for developing HIVAN in people of African ancestry. Nonetheless, other endogenous factors are needed, as well, because people of African ancestry who do not carry the APOL1 risk variants, and HIV-transgenic (Tg) mice, also develop HIVAN. During the past year, Dr. Ray’s HIV research program, which is supported by three NIH R01 grants, found that HIV-1can infect podocytes cultured from the urine of HIV-positive children carrying the APOL-1 G1 risk alleles and induce the expression of APOL-1 in these cells. In this manner, HIV-1 can induce the processes of autophagy and/or cell death in these cells, leading to HIVAN. In addition, they have found that HIV- induced activation of the Rho-A pathway plays a critical role in this process, and they have developed an assay to identify circulating factors present in the urine of HIV-positive children that are capable of inducing the activity of Rho-A in cultured podocytes and renal glomerular endothelial cells. This assay could be used to follow the outcome of other pediatric renal diseases. In addition, the program made progress in assessing the clinical value of a new panel of urinary biomarkers that, in combination with the APOL1 genotype, may allow diagnosis of HIVAN in children without performing a renal biopsy.
Genetic Models Using Drosophila for Heart and Kidney Disease
- Zhe Han, Ph.D.
- Yulong Fu, Ph.D.
- Pei Wen, Ph.D.
- Jin-gu Lee, Ph.D.
Dr. Han’s lab develops genetic disease models using Drosophila with a focus on heart, kidney and blood diseases. He has developed a new approach to generate personalized Drosophila models carrying the same mutations as patients and uses these fly models to study disease mechanism and develop precision medicine treatments based on the disease mechanism. Dr. Han’s lab combines the power of cutting-edge genetic tools in Drosophila with patient exome sequencing and advanced Bioinformatic analysis and is funded by multiple NIH R01 grants. The lab was the first in the world to develop Drosophila model for Nephrotic Syndrome (NS), a collection of genetic kidney diseases that affect the glomerular kidney filtration. The lab showed that most of the 40 known genes involved in NS have conserved fly homologs that are essential for the function of the nephrocyte, a specialized filtration cell type in Drosophila. Dr. Han’s lab is currently building new NS models to study molecular disease mechanisms, as well as to test potential therapeutic drug treatments.
Genetic variants in the APOL1 gene is the cause of the much higher risk of kidney diseases in African Americans, but the mechanism is not clear. The lab developed the first APOL1 nephropathy model in Drosophila nephrocytes, and discovered that APOL1 affects organelle acidification, nephrocyte hypertrophy, and accelerated cell death. Using a Drosophila genetic screen approach, The lab has identified novel cofactors for APOL1, which are required for APOL1 toxicity. Using RNA-seq, Dr. Han’s group also identified downstream factors for APOL1. These novel findings could be used to develop targeted therapeutic treatments for APOL1 nephropathy.
HIV nephropathy is a major complication for HIV patients who have received treatment to control proliferation of the virus. The Han lab, in collaboration with Dr. Ray, is developing a Drosophila HIV-1 nephropathy models to study the molecular mechanisms of HIV-associated nephropathy.
Kidney Fibrosis
- Zhe Han, Ph.D.
- Jun-yi Zhu, Ph.D.
Fibrosis, the process of scarring, is a common mechanism by which kidney injury progresses to kidney loss. In collaboration with Bill Schnaper, M.D., at Northwestern University, Dr. Han discovered that a signaling molecule, Smad Anchor for Receptor Activation, or SARA, might maintain kidneys in a healthy state by preventing them from producing scar when they are damaged. The goal of this project is to better understand the way SARA acts in the tissues to oppose scarring, to learn what makes SARA levels go up or down, and to identify what the actions of SARA are that prevent scarring, using Drosophila and mouse as model systems. This project is currently funded by an NIH R01 grant.
Clinical Aspects of Pediatric Kidney Disease
- Patricio Ray, M.D.
- Asha Moudgil, M.D.
- Jharna Das, Ph.D.
Dr. Ray’s group, in collaboration with Drs. An Massaro, Mary Revenis, Charu Gupta and Sofia Perazzo, from the Divisions of Neonatology and Nephrology, developed a new approach to assess the renal outcome of term newborns with hypoxic ischemic encephalopathy (HIE). This approach includes the development of a new definition of acute kidney injury (AKI) for newborns during the first week of life. These findings are helping neonatologists identify newborns with AKI during the 1st days of life. Furthermore, this new definition, will facilitate the discovery and testing of more sensitive urinary biomarkers of AKI during the first week of life. Finally, Drs. Ray and Das, in collaboration with Dr. Moudgil, developed a new bioassay to identify glomerular changes induced by permeability factors released into the circulation of children with nephrotic syndrome, and focal segmental glomerulsclerosis (FSGS) recurring after renal transplantation.
Cardiovascular Disorders
Congenital Heart Disease
- Zhe Han, Ph.D.
- Jun-yi Zhu, Ph.D.
- Hanhan Liu, Ph.D.
Mutations in histone-modifying genes have been identified as a major risk factors from the genomic sequencing of Congenital Heart Disease (CHD) patients, but the roles of histone modifying genes in heart development remain unclear. The goal of this project is to study the role of histone-modifying genes in heart development, using Drosophila as a model system. The team will also establish a series of fly models of CHD, using the exact same histone modifying gene mutations identified from the patients, to better understand the disease mechanisms.
Heparin-Binding Growth Factors
- Patricio E. Ray, M.D.
- Jharna Das, Ph.D.
- Aswini K. Panigrahi
- Elizabeth Wilson, M.D.
- Anthony Sochet, M.D.
- John Berger, M.D.
- Pingtao Tang, Ph.D.
Critically ill children treated with heparin during extracorporeal membrane oxygenation (ECMO) and cardiopulmonary bypass (CPB) are at high risk of developing severe capillary leak syndromes, excessive bleeding, and acute kidney injury. These events are attributed to multifactorial causes, including inflammatory cytokines and the anti-coagulant activity of heparin. However, very little is known about how heparin-binding growth factors released into the circulation of critically ill children modulate this process. A nonsurgical intervention that can effectively control post-operative vascular leakage and bleeding is needed. This year, a team of investigators from Children’s lead by Dr. Ray (Gen Med) and Drs. Wilson, Sochet Berger (Division of Cardiac Intensive Care), discovered that the urinary levels of Fibroblast Growth Factor-2 (FGF-2) and Vascular Endothelial Cell Growth Factor-A (VEGF-A), can be used to identify children at high risk of developing severe bleeding complications and vascular leakage after CPB. This work, supported by an R01 grant from the NHLBI provides significant clinical evidence to validate the studies done in Dr. Ray’s laboratory showing that FGF-2 release into the circulation of mice treated with heparin-like drugs precipitates the development of severe bleeding complications. Furthermore, additional work done in this lab, found that Angiopoietin-1, an anti-permeability/anti-inflammatory growth factor, prevented lethal bleeding complications in mice treated with FGF-2 and heparin-like drugs, without normalizing their anticoagulant status. These findings were published in the American Journal of Physiology (Heart and Circulation Physiology). Thus, urinary levels of FGF-2 and VEGF-A might become reliable biomarkers to identify critically ill children at high risk of bleeding when subjected to CPB. In addition, a team of investigators lead by Drs. Das and Panigrahi, are currently developing new techniques to identify additional vascular permeability factors released into the circulation of children treated with heparin.
Bone Health Program
- Laura L. Tosi, M.D.
Peak bone mass is reached by early adulthood and serves as a person’s bone bank for the rest of their life. The more bone that is deposited during a person’s childhood and adolescence, the longer it will last as that person grows older. Good bone deposits made when a child is young may help prevent osteoporosis in adulthood. Unfortunately, many children, particularly those with a genetic susceptibility to low bone density and/or with chronic medical conditions that require medications that are toxic to bone, are at risk for frequent fractures. The Bone Health Program seeks to maximize the bone health of children and adults who are at increased risk for fractures by providing multidisciplinary diagnosis and treatment services for them, as well as the opportunity to participate in innovative research.
Brittle Bones Disease Rare Disease Clinical Research Consortium (BBD-RDCRC)
The Bone Health Program is a member of the Brittle Bones Disorders Rare Disease Clinical Research Consortium (BBD-RDCRC) within the Rare Disease Clinical Research Network funded by the NIH. The consortium’s goal is to perform collaborative clinical research on brittle bone disorders, including a longitudinal observational study driven by genotypic association. Dr. Tosi is the site PI for this program. She is also co-PI of a pilot project that seeks to explore the use of the PROMIS tool to provide valid quality of life (QOL) measures in individuals with osteogenesis imperfecta (OI). This work has been motivated by the fact that all current outcome measures in OI have been developed by medical experts without input from patients, and patients and clinicians often disagree about the level of disease burden. The project seeks to develop clinical scoring instruments that capture the disease characteristics of importance to individuals with OI in order to fully compare and contrast the impact of new treatments, as well as to determine future needs and research topics.
Defining Genetic Influences on Bone and Muscle Strength and Development Projects
Dr. Tosi has had the opportunity to partner with members of the National Human Genome Research Institute for more than 15 years as part of their effort to identify the cause of mosaic limb overgrowth. This work encouraged her interest in rare skeletal disorders and led to her focusing on these disorders in her research program. In addition, the Bone Health Program hosts a summer research program for medical students interested in a career in musculoskeletal health that focuses on discovering and validating genetic variants associated with musculoskeletal health and disease.
Novel Genomic Technologies
Novel Genomic Technologies
- Eric Vilain, M.D., Ph.D.
- Hayk Barseghyan, Ph.D.
- Surajit Battacharaya, Ph.D.
- Emmanuèle Délot, Ph.D.
To search for variants in the about 98.5 percent of the genome not directly encoding exons, the team uses Whole Genome Sequencing (WGS), with the goal of validating this research technology for clinical use. Through the support of the Gabriella Miller Kids First initiative of the NIH, the team has been invited to help develop the new Cavatica platform the NIH is creating as part of their new Pediatric Data Resource center, as well as to collaborate in beta testing the Broad Institute’s seqr platform, developed for variant calling in WGS for rare disorders.
As the short-read technology of WGS limits its ability to identify large structural variants, the team has explored optical Whole Genome Mapping (WGM), a novel technology where ultra-long molecules of DNA, fluorescently labeled at known intervals, are pushed through nanochannels to read the pattern of fluorescence on single molecules. Breaks in the pattern signal the presence of structural variants. Children’s National scientists demonstrated that this platform could be used to detect variants in patients with Duchenne Muscular Dystrophy, with higher precision for breakpoint localization than current methods (Barseghyan et al. 2017). WGM (also called Next-Generation Mapping) was able to detect insertions smaller than what is currently reported with clinical chromosomal microarrays, deletions from 45 to 250 kb in hemizygous or heterozygous states, as well as a large balanced inversion of 5.1 Mb, which would not have been detected by chromosomal microarray and had taken months to demonstrate with traditional methods. WGM is poised for use in clinical diagnosis and will now apply this technology to search for new etiology in conditions affecting sexual and reproductive development and other undiagnosed rare diseases.
The team has acquired new equipment to complement these studies with long-read sequencing technology. The platform (developed by Oxford Nanopore) detects current variation when a molecule passes through a pore, allowing sequencing of DNA or RNA, even in repetitive regions. It can detect sequence variants as well as structural variants, and potentially epigenetic modifications simultaneously, a disease etiology currently difficult to assess.
To improve its ability to meaningfully interpret these genome-wide searches for rare disease etiology, the team is designing new software to integrate the data sets obtained by the various analytic methods (WGS long/short reads, WGM, exome, RNASeq), as well as software to improve variant calling for structural variants, a field with current serious limitations. This will be developed in collaboration with the new CRI Bioinformatics Unit.
Bioinformatics
- Hiroki Morizono, Ph.D.
- Eric Vilain, M.D., Ph.D.
- Susan Knoblach, Ph.D.
- Wei Li, Ph.D.
- Kazue Hashimoto-Torii, Ph.D.
The CRI Bioinformatics Unit is a new initiative sponsored by the Center for Genetic Medicine Research, the Center for Translational Science, The Clinical and Translational Science Institute at Children’s National (CTSI-CN), the District of Columbia Intellectual and Developmental Disabilities Research Center (DC-IDDRC) and the Chief Research Officer’s office. The objective is to create a research home for bioinformaticians and computational biologists drawn from several CRI centers, and to provide bioinformatics services for investigators at Children’s National through the creation of a Bioinformatics core. The unit will develop, implement and disseminate tools and technologies for working with Omics data, while the Bioinformatics Core will provide CRI investigators with analytic support for their informatics projects. Dr. Li is a new faculty recruit in the Center for Genetic Medicine Research who brings an extensive background in modeling CRISPR/CAS9 targeting. Through the CTSI-CN, a partnership has been established with bioinformatics groups at GW to promote the formation of a regional collaboratorive where the next generation of biomedical researchers will be trained in data science and quantitative biology.
Genomics
- Susan Knoblach, Ph.D.
The Genomics Core continues to provide access to state-of-the-art equipment and expertise for investigators interested in using genomic approaches in their research. The arrival of Dr. Vilain as the new director for the Center of Genetic Medicine, brought new equipment and technologies to the core, including a BioNano Optical Genomics Scanner. This instrument detects large structural variations in gene sequence more reliably and at lower cost than other standard sequencing methods. It is particularly relevant for Children’s National, because large structural variations are responsible for many childhood diseases and conditions, including cancers and developmental disorders. Dr. Vilain has also purchased MinION and GridION sequencers that the core may use to sequence very long regions of RNA/DNA, as well as to detect modifications in these nucleic acids that affect how cells use them. A third new technology is single-cell RNA sequencing---this method is being developed to sequence RNA from different cell types that normally reside together in a particular tissue. This approach can provide new contextual insight(s) about cellular processes and pathways.
Proteomics
- Aswini K. Panigrahi, Ph.D.
Proteomics continue to play critical roles in the research with advanced technological platforms, and bringing together clinicians and basic scientists. Over the past year, the core supported investigators in different areas of research, including pediatric brain tumors, pediatric pharmacology, pharmacodynamics biomarkers, muscular dystrophies, urea cycle disorder, asthma, and neurodegenerative diseases. This provided new insights into molecular mechanism of pediatric diseases and helped develop new therapeutic targets. Using proteo-genomic approaches potential new antigens were identified in brain tumor tissues (collaborations with Dr. Rood). The team developed a targeted proteomic assay for biomarker validation in DMD and IBD sera samples (collaborations with Dr. Conklin).
Imaging Technologies
- Jyoti Jaiswal, Ph.D.
- Stanley Fricke, Ph.D.
The CRI Light Microscopy and Image Analysis Core is partially supported by the National Institute of Child Health and Human Development (NICHD)/NIH-supported U54 grant for the DC-IDDRC (Intellectual and Developmental Disabilities Research Center in the District of Columbia metropolitan area). This core, directed by Dr. Jaiswal, provides a broad array of advanced microscopy equipment allowing access, advice and training to investigators at CRI and in the DC-IDDRC for research involving microscopic imaging. The core is managed by Shivaprasad Bhuvanendran, M.S., and has been pushing ahead with development and use of superresolution microscopy and high resolution deep tissue imaging. The core supports over 40 federally funded research projects and helps develop new research projects, six of which received federal funding this year and have facilitated trainees to start their own laboratories on childhood-related research. The core also organizes workshops and vendor-provided trainings to help investigators access new technologies and meet the growing need for high and super-resolution microscopic imaging.
The NIH National Heart, Lung, and Blood Institute funds Dr. Fricke’s work to diagnose and treat cardiovascular and lung disease in children by creating high resolution, ultrafast systems for MRI (magnetic resonance imaging). Demonstrating a 128,000-fold gain in slew rate, the lab’s work promises to reduce MRI exam sessions from the current one hour to a few minutes, potentially eliminating the need for anesthesia in young children and permitting stop-motion analysis for cardiac studies. Dr. Fricke is developing multimodality preclinical imaging technology for placing nanoparticles in cells, tracking their movement in the body, and locating them for biopsy. Working with Dr. Paul Wang from Howard University, and supported by an NIH award, they have setup a Bruker 7 T (300 MHz) AVANCE III NMR/MRI machine at Howard University animal imaging facility. This machine has a 210 mm bore size magnet capable of conducting high resolution imaging and spectroscopy studies in larger animals (up to piglets). It accommodates various imaging and spectroscopy studies and is equipped with an animal monitoring system.
Biostatistics Support and Collaborations
- Heather Gordish-Dressman, Ph.D.
After serving as the primary biostatistician for the Center for Genetic Medicine Research, Dr. Gordish-Dressman has transitioned to the Center for Translational Science. She continues to provide statistical support for researchers within the center and to others outside the center, including other institutions. Internal collaborations during 2017 included several projects with the Department of Anesthesiology on the effectiveness of the perioperative surgical home instituted at Children’s National and a long-term collaboration with Dr. Tosi researching the effects of genetic modifiers on muscle and bone phenotypes. A large portion of her work is in assessing the natural history of both Duchenne and Becker Muscular dystrophy and has led to several publications including a seminal paper in the Lancet. External collaborations include a project investigating the mdx mouse model of disease in collaboration with multiple academic centers throughout the U.S. and Europe.
Education
The Genomics and Bioinformatics Ph.D. Program, Institute for Biomedical Sciences, the George Washington University.
Director: Eric Vilain, M.D., Ph.D.
Co-directors and Program Advisors: Ljubica Caldovic, Ph.D., Keith Crandall, Ph.D., Raja Mazumder, Ph.D.
The Center for Genetic Medicine Research is home to the George Washington University Department of Genomics and Precision Medicine (GPM) within the School of Medicine and Health Sciences. Center faculty account for most GPM members. The Genomics and Bioinformatics Graduate Program aims to prepare students to become the next generation of biomedical scientists. The students pursue their dissertation research in laboratories at the Children’s National Hospital, GW and the NIH.
Relatively new on the university scene, the center has carved out a popular set of classes, including Molecular Bases of Human Diseases, Genes to Cells, Introduction to Genomics, Proteomics and Bioinformatics, Applied Biostatistics, and the combined Genetic Medicine and Integrative Systems Biology Seminars. The center’s faculty members teach additional graduate courses and medical student and physician assistant courses at GW.
Selected Publications from 17/18
- Barseghyan H, Tang W, Wang RT, Almalvez M, Segura E, Bramble MS, Lipson A, Douine ED, Lee H, EC Délot, Nelson SF, Vilain E. (2017) Next-Generation Mapping (NGM): A novel approach for detection of pathogenic structural variants with a potential utility in clinical diagnosis. Genome Medicine 9(1). PMID: 29070057.
- Defour A, Medikayala S, Van der Meulen JH, Hogarth MW, Holdreith N, Malatras A, Duddy W, Boehler J, Nagaraju K, Jaiswal JK (2017) Annexin A2 links poor myofiber repair with inflammation and adipogenic replacement of the injured muscle. Hum. Mol. Genet. 26:1979-91.
- Délot EC, Papp JC, the DSD-TRN Genetics Workgroup, Sandberg DE, Vilain E. 2017. Genetic Diagnosis of Disorders of Sex Development (DSD): the DSD-Translational Research Network experience. Endocrinol. Metab. Clin. N. Am. 46:519-537.
- Hasegawa K, Pérez-Losada M, Hoptay CE, Epstein S, Mansbach JM, Teach SJ, Piedra PA, Camargo Jr. CA, Freishtat RJ (2018) RSV vs. rhinovirus bronchiolitis: difference in nasal airway microRNA profiles and NFkB signaling. Pedatric Research 83: 606-614.
- Hoffman EEP, Riddle V, Siegler MA, Dickerson D, Backonja M, Kramer WG, Nagaraju K, Gordish-Dressman H, Damsker JM, McCall JM (2018) Phase 1 trial of vamorolone, a first-in-class steroid, shows improvements in side effects via biomarkers bridged to clinical outcomes. Steroids 134:43-52.
- Horn A, Van der Meulen JH, Defour A, Hogarth M, Sreetama SC, Reed A, Scheffer L, Chandel NS, Jaiswal JK. (2017) Mitochondrial redox signaling enables repair of injured skeletal muscle cells. Sci Signal. 2017 Sep 5;10(495). pii: eaaj1978. doi: 10.1126/scisignal.aaj1978. (Cover of the 9 September 2017 issue)
- Jang YJ, LaBella AL, Feeney TP, Braverman N, Tuchman M, Morizono H, Ah Mew N, Caldovic L (2018) Disease-causing mutations in the promoter and enhancer of the ornithine transcarbamylase gene. Human Mutation 39:527-36.
- Kirmse B, Cabrerra-Luque J, Ayyub O, Cusmano K, Chapman K, Summar M. (2017) Plasma fibroblast growth factor-21 levels in patients with inborn errors of metabolism. Mol. Genet. Metab. Rep. 13:52-54.
- Novak JS, Hogarth MW, Boehler JF, Nearing M, Vila MC, Heredia R, Fiorillo AA, Zhang A Hathout Y, Hoffman EP, Jaiswal JK, Nagaraju K, Cirak S, Partridge TA (2017) Myoblasts and macrophages are required for therapeutic morpholino antisense oligonucleotide delivery to dystrophic muscle. Nature Communications. 8(941):1-13.
- Wells E, Kambhampati M, Damsker JM, Gordish-Dressman H, Yadavilli S, Becher OJ, Gittens J, Stampar M, Packer RJ, Nazarian J. (2017) Vamorolone, a dissociative steroidal compound, reduces pro-inflammatory cytokine expression in glioma cells and increases activity and survival in a murine model of cortical tumor. Oncotarget. 8(6):9366-9374.
- Zhu JY, Fu Y, Richman A, Zhao Z, Ray PE, Han Z (2017) A personalized Drosophila model of COQ2 nephropathy rescued by the wild-type human COQ2 allele and dietary Q10 supplementation. J. Am. Soc. Nephrol. 28(9):2607-2617. PMCID: PMC5576924. (Featured on the cover of JASN for the September 2017 issue).
- Fiorillo AA, Tully CB, Damsker JM, Nagaraju K, Hoffman EP, Heier CR. Muscle miRNAome shows suppression of chronic inflammatory miRNAs with both prednisone and vamorolone. Physiol Genomics. 2018 Jun 8. doi: 10.1152/physiolgenomics.00134.2017. [Epub ahead of print]